News Release
Researchers at UC San Diego Unveil New Interactive Web Tool for Visualizing Large-Scale Phylogenetic Trees
March 17, 2021
The proliferation of sequencing technologies and an ever-increasing focus on ‘omic studies over the past several years have spawned massive quantities of data and many tools to process and analyze the results. As these tools have evolved, so has the need for even greater scale and the ability to visualize complex data sets in more efficient ways.
Born of these needs for advancement, a team of researchers with the Center for Microbiome Innovation (CMI) at the University of California San Diego (UC San Diego), led by Kalen Cantrell and Marcus W. Fedarko, unveiled EMPress, a new interactive visualization tool, in an article published in mSystems on March 16, 2021. The paper, entitled “EMPress Enables Tree-Guided, Interactive, and Exploratory Analyses of Multi-omic Data Sets,” showcases the tool’s functionality, versatility, and scalability, and explores some of the potential applications in visualization and data analysis.
"Although there are many excellent existing tools for visualizing trees, we wanted to create a tool that could be used within a web browser and could scale to really large trees," Fedarko remarked when discussing the development process. "Another motivation was that there were many custom features that no existing tool, to our knowledge, supported - for example, the ability to interactively link samples in an ordination with a phylogenetic tree, showing the sequences contained within each sample, or going the opposite way, the samples containing a given sequence."
Aside from interactively integrating ordinations with trees, EMPress also introduces the ability to animate a tree’s changes over time. While other tools have included a temporal dimension to trees, the ability to animate the tree alongside an ordination is a novel feature that the team believes will enhance the exploratory analysis of ‘omic data sets.
What began as an undergraduate project at UC San Diego’s Early Research Scholars Program has since grown into a collaboration that spans both academia and industry. The team at CMI has worked closely with IBM Research’s Artificial Intelligence for Healthy Living Center throughout the process, as well as researchers at the Simons Foundation, Cornell University, and Justus Liebig University.
Developing a tool that was not only web-based but capable of storing, processing, and visualizing such large phylogenetic trees presented numerous challenges. As the scale of microbiome and other ‘omic studies has continued to grow, so too has the technical complexity required to accommodate and analyze the large, complex data sets those studies produce.
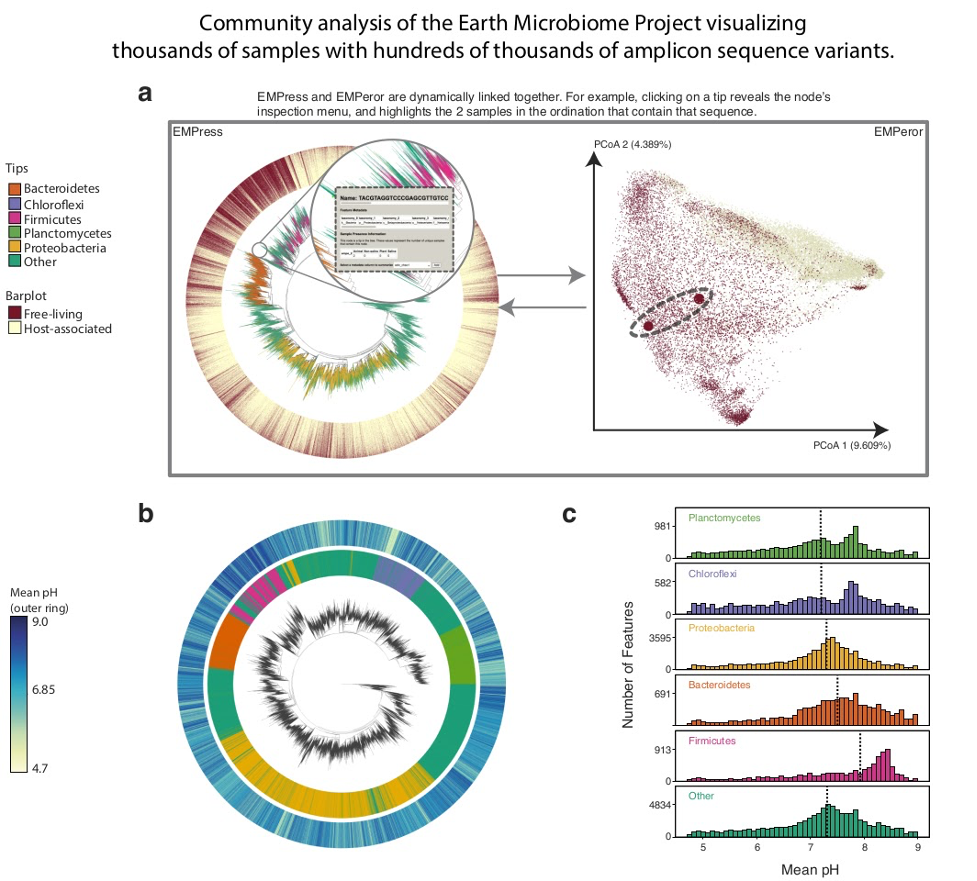 |
|
FIG 1 Earth Microbiome Project paired phylogenetic tree (including 756,377 nodes) and unweighted UniFrac ordination (including 26,035 samples). (a) Graphical depiction of EMPress’ unified interface with fragment insertion tree (left) and unweighted UniFrac sample ordination (right). Tips are colored by their phylum-level taxonomic assignment; the barplot layer is a stacked barplot describing the proportions of samples containing each tip summarized by level 1 of the EMP ontology. Inset shows summarized sample information for a selected feature. The ordination highlights the two samples containing the tip selected in the tree enlarged to show their location. (b) Subset of EMP samples with pH information: the inner barplot ring shows the phylum-level taxonomic assignment, and the outer barplot ring represents the mean pH of all the samples where each tip was observed. (c) pH distributions summarized by phylum-level assignment with median pH indicated by dotted lines. Interactive figures can be accessed at https://github.com/knightlab-analyses/empress-analyses. |
"We resolved some of these issues by creating our own custom WebGL, shaders which are capable of visualizing millions of points, and implementing high-performance data structures such as the balanced parentheses tree that helped drastically reduce the memory footprint of EMPress," according to Cantrell. As the code has been refined and optimized through several phases of development, it has enabled the analysis of larger and larger data sets, including a tree with over 750,000 nodes.
While the article in mSystems represents the tool’s formal unveiling, it has already seen use in several studies and applications. Researchers at the CMI utilized EMPress in their recently published analysis of the composition and microbial load of the human oral microbiome. Additionally, epidemiologists with UC San Diego’s Return to Learn program have been using the tool to help manage the university’s response to the COVID-19 pandemic. These applications have provided critical feedback and facilitated improvements to continue streamlining the analyses for which EMPress is designed.
As development continues, there are several planned features, including the ability to explore time-series data, potential anomaly detection, implementing support for annotating various types of data, and further improving performance to allow for larger data sets. The tool’s source code is available on GitHub, and the team is eager to see how their peers expand upon their work.
“Just as EMPress builds on the many existing tree visualization tools in the literature (iTOL, PHYLOViZ, Anvi'o, SigTree, FigTree, ggtree, TopiaryExplorer, MicroReact, Archaeopteryx, etc.), we're sure that other tools will pop up soon enough that build on the concepts used in EMPress -- we look forward to seeing how the landscape of these tools changes in the future,” Fedarko added.
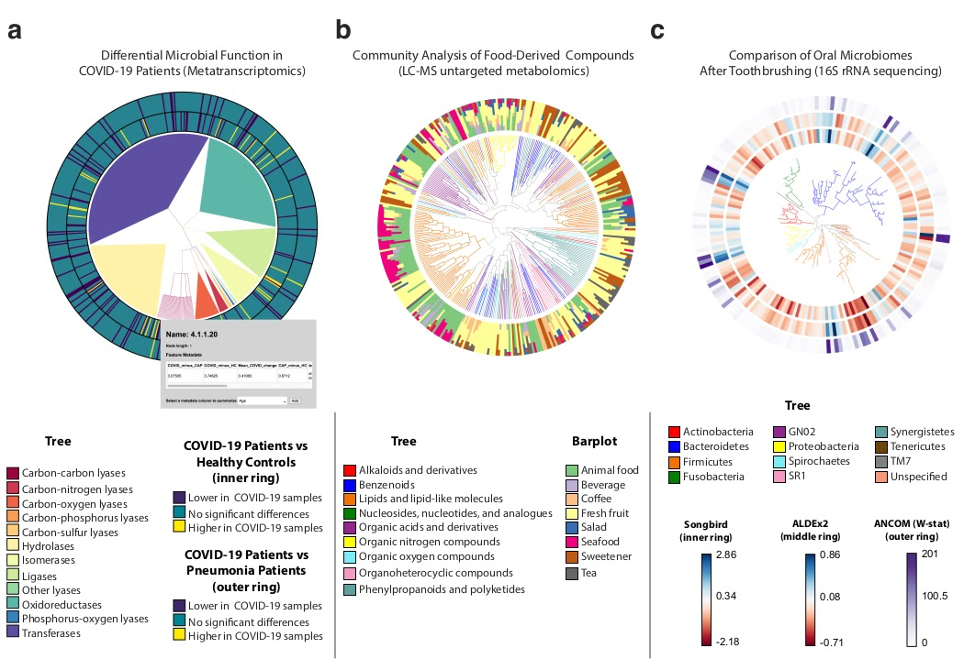 |
|
FIG 2 EMPress is a versatile exploratory analysis tool adaptable to various ‘omics data types. (a) RoDEO differential abundance of microbial functions from metatranscriptomic sequencing of COVID-19 patients (n = 8) versus community-acquired pneumonia patients (n = 25) and versus healthy control subjects (n = 20). The tree represents the four-level hierarchy of the KEGG enzyme codes. The barplot depicts significantly differentially abundant features (P , 0.05) in COVID-19 patients. Clicking on a tip produces a pop-up insert tabulating the name of the feature, its hierarchical ranks, and any feature annotations. (b) Global FoodOmics Project LC-MS data. Stacked barplots indicate the proportions of samples (n = 70) (stratified by food) containing the tips in an LC-MS Qemistree of food-associated compounds, with tip nodes colored by their chemical superclass. (c) De novo tree constructed from 16S rRNA sequencing data from 32 oral microbiome samples. Samples were taken before (n = 16) and after (n = 16) subjects (n = 10) brushed their teeth; each barplot layer represents a different differential abundance method’s measure of change between before- and after-brushing samples. The innermost layer shows estimated log-fold changes produced by Songbird, the middle layer shows effect sizes produced by ALDEx2, and the outermost layer shows the W-statistic values produced by ANCOM (see Materials and Methods). The tree is colored by tip nodes’ phylum-level taxonomic classifications. Interactive figures can be accessed at https://github.com/knightlab-analyses/empress-analyses. |
Additional co-authors include Gibraan Rahman, Daniel McDonald, Yimeng Yang, Thant Zaw, Antonio Gonzalez, Mehrbod Estaki, Qiyun Zhu, Erfan Sayyari, George Armstrong, Anupriya Tripathi, Julia M. Gauglitz, Clarisse Marotz, Nathaniel L. Matteson, Cameron Martino, Se Jin Song, Austin D. Swafford, Pieter C. Dorrestein, Kristian G. Andersen, Yoshiki Vázquez-Baeza, and Rob Knight, all at UC San Diego; Stefan Janssen at Justus Liebig University; Niina Haiminen and Laxmi Parida at the IBM Thomas J. Watson Research Center; Kristen L. Beck and Ho-Cheol Kim at the IBM Almaden Research Center; James T. Morton at the Simons Foundation; Jon G. Sanders at Cornell University; and Anna Paola Carrieri at IBM Research Europe.
This piece was written by CMI’s contributing editor Cassidy Symons
Media Contacts
Brittanie Collinsworth
UC San Diego Center for Microbiome Innovation
858-534-8390
b4collinsworth@ucsd.edu